
 |
Александр Пилипенко «Новости эволюции человечества» Посмотреть видео |
 |
Ася Василовская «Эволюция современного искусства» Посмотреть видео |
 |
Яна Степанчук «Генетическое тестирование преимлантационных эмбрионов» Посмотреть видео |
 |
Марина Шарапова «Биоэтика: что, где, зачем» Посмотреть видео |
 |
Даниил Юхтанов «Сложная анатомия простейших» Посмотреть видео |
 |
Артём Нурисламов «Центромеры» Посмотреть видео |
 |
Эвелина Кабирова «Энхансеры в пространстве и времени» Посмотреть видео |
 |
День Дарвина 2024 Эволюция теорий об эволюции(лекция) Посмотреть видео |
 |
День Дарвина 2024 Интервью с Дарвином(блокбастер) Посмотреть видео |
 |
П.Бородин «Инадаптивная эволюция Владимира Ковалевского» Посмотреть видео |
 |
Ольга Посух «Селективное преимущество гетерозигот Посмотреть видео |
 |
Ирина Соколова «Растения в стрессе» Посмотреть видео |
 |
Вениамин Фишман, Нариман Баттулин и др. «Лайфхаки: Как использовать ChatGPT с пользой для науки» Посмотреть видео |
 |
Артемий Иванов «Куда нам расти в защите растений» Посмотреть видео |
 |
Григорий Степанов «Модификации нуклеотидов РНК и регуляция экспрессии генов» Посмотреть видео |
 |
Олег Костерин «При царе Горохе: ареал, местообитания и популяции дикого гороха» Посмотреть видео |
 |
Владимир Науменко «Флуоресцентные белки в нейробиологии» Посмотреть видео |
 |
Анастасия Чвилева «HARs: участки генома, которые сделали нас людьми» Посмотреть видео |
 |
Людмила Науменко «Простейшие на пути к многоклеточности» Посмотреть видео |
 |
Все Нобелевские премии 2023 в одной лекции Посмотреть видео |
 |
П.Бородин «Жизнь и приключения Альфреда Рассела Уоллеса в Бразилии, Ост-Индии и в мире духов» Посмотреть видео |
 |
Татьяна Шнайдер «Ксенотрансплантация» Посмотреть видео |
 |
Александр Пилипенко «Новости антропогенеза» Посмотреть видео |
 |
Александр Злобин «Селекция животных сквозь призму генетики» Посмотреть видео |
 |
Фёдор Абрашитов Как вырастить исследователя Посмотреть видео |
 |
Нариман Баттулин Как устроена наука. История одного научного факта: от открытия до закрытия. Загрузить презентацию Посмотреть видео |
 |
Екатерина Хогоева (НГУ) «Планета Земля и геофизические поля» Посмотреть видео |
 |
Дмитрий Жарков, Павел Бородин, Вениамин Фишман, Юлия Бокова и другие «Лайфхаки: как писать заявки на гранты и отчеты по ним» Посмотреть видео |
 |
Александра Зубкова «Пауза в отношениях, или транскрипционный стресс РНК-полимеразы II» Посмотреть видео |
 |
Илья Жданков «Баба Яга от неолита до метамодерна: эволюционный анализ фольклорных сюжетов» Посмотреть видео |
 |
«Лайфхаки генной инженерии» Посмотреть видео |
 |
День Дарвина 2023 Посмотреть видео |
 |
День Дарвина 2023 1. Минилекция Софии Хасиной «Старые девы, кошки, мыши, шмели и экология по Дарвину» Посмотреть видео |
 |
День Дарвина 2023 2. Трагедия в стихах Фёдора Абрашитова в исполнении Елены Кирилловой, Дениса Матвеева, Ивана Федорищенко, Богдана Титова, Андрея Сафина, Екатерины Дорогановой, Анастасии Мосеенковой, Эммы Мельниковой Читать |
 |
День Дарвина 2023 3. Премьера фильма «Возвращение Дарвина (2023)» Посмотреть видео |
 |
Владимир Трифонов «Экстрахромосомная ДНК: неуловимая часть генома» Посмотреть видео |
 |
Артём Нурисламов «О теломерах: твёрдо и чётко» Посмотреть видео |
 |
Илья Бетеров (ИФП СОРАН) «Квантовый компьютер на холодных атомах» Посмотреть видео |
 |
Антон Цыбко «Тайная жизнь серотониновых рецепторов» Посмотреть видео |
 |
Даниил Юхтанов «Увидеть митохондрию и …» Посмотреть видео |
 |
Александр Пилипенко (ИЦИГ) Илья Бетеров (ИФП) Евгений Мостович (НГУ) Иван Полторацкий(НГУ) Дмитрий Березняков (НГУ) Екатерина Исупова (ИЭОПП) Все Нобелевские премии 2022 г. в одной лекции Посмотреть видео |
 |
Сергей Кулемзин «Клеточные инструменты терапии рака» Посмотреть видео |
 |
Наталия Рудая «Микрофоссилии и реконструкция прошлого Земли» Посмотреть видео |
 |
Владимир Музыка «Пути и механизмы специализации нейронов» Посмотреть видео |
 |
Светлана Модина «Геном мамонта»» Посмотреть видео |
 |
Павел Сальников «Запутанная история о функциях хроматиновых петель» Посмотреть видео |
 |
Дмитрий Щербаков «Реконструкция эволюции морфологических признаков» Посмотреть видео |
 |
Дмитрий Щербаков «Следы отбора в некодирующих районах генома». Посмотреть видео |
 |
День рождения Грегора Менделя в ИЦИГ Татьяна Шнайдер «Похвальное слово Менделю» (3.35 -14.05) Эмиль Весна «Дорога плуга и креста» (поэма — 18.15 -19.40) Дмитрий Щербаков «»Следы отбора в некодирующих районах генома»(лекция -20.40-1.20.29). Посмотреть видео |
 |
Нариман Баттулин «Лекция про жесткость генома» Посмотреть видео |
 |
Александр Пилипенко «Новости эволюции человечества» Посмотреть видео |
 |
Проф. О.А.Донских (НГУ) «Наука и религия: история взаимоотношений»» Посмотреть видео |
 |
Степан Белякин «Ветеринарная геномика» Посмотреть видео |
 |
Яков Цепилов «Генетика продолжительности жизни» Посмотреть видео |
 |
Анна Гурина (ИСИЭЖ) «Насекомые позднего плейстоцена» Посмотреть видео |
 |
Наталья Кох «Противоречия доказательной и персонализированной медицины» Посмотреть видео |
 |
Елена Уфимцева «»Микобактерия туберкулеза и человек – парадоксы сосуществования» Посмотреть видео |
 |
«День Дарвина 2022 Илья Жданков «Как можно размножаться? Павел Никулин «Восьмая ода Дарвину» Премьера психологического триллера «Премия Дарвина 2022» Посмотреть видео |
 |
Антон Цыбко «Эволюция и нейробиология любви» Посмотреть видео |
 |
Сергей Кулемзин «Нейтрализующие антитела к SARS-CoV-2» Посмотреть видео |
 |
Елизавета Куликова «Бежать, думать, эволюционировать» Посмотреть видео |
 |
Dr. Francisco J. Ruiz-Ruano (University of East Anglia) Satellite DNA: a toolbox for cytogenomics Посмотреть видео |
 |
Dr. Alexander Suh (University of East Anglia) Programmed DNA elimination Посмотреть видео |
 |
Павел Сальников «Генетика развития конечностей». Посмотреть видео |
 |
Татьяна Бикчурина «Мейоз и гибридная стерильность». Посмотреть видео |
 |
Маргарита Романенко «Ре-вакцинация: когда, кому, зачем» Посмотреть видео |
 |
Алексей Дорошков «Транспорт в растениях или как деревья обманывают законы физики». Посмотреть видео |
 |
Все Нобелевские премии 2021 в одной Лекции Посмотреть видео |
 |
Вениамин Фишман «Геномика одиночных клеток» Посмотреть видео |
 |
Ася Хрущева «Есть ли жизнь в современной школе? Посмотреть видео |
 |
Всеволод Ефременко (ИНГГ СО РАН) «Биотические кризисы Мелового периода» Посмотреть видео |
 |
Дмитрий Долгушин «Настанет год, России черный год…: холерная эпидемия 1830— 1831 гг. в жизни и литературе» Посмотреть видео |
 |
Алла Красикова «Не только генетический код: Функции некодирующей белки РНК» Посмотреть видео |
 |
Дмитрий Гаськов «Таргетная и иммунотерапия в онкологии: что это и как это работает» Посмотреть видео |
 |
Василий Марусин «Кембрийский взрыв и агрономическая революция» Посмотреть видео |
 |
Александр Пилипенко «Новости эволюции человечества» Посмотреть видео |
 |
Елена Убогоева «Секрет долголетия растений» Посмотреть видео |
 |
Софья Герасимова «Молекулярный календарь огородника» Посмотреть видео |
 |
Александра Клименко «Эволюция ролевых игр живого действия» Посмотреть видео |
 |
Денис Ларкин «Конвергентная эволюция коров на полюсе холода» Посмотреть видео |
 |
Алексей Дорошков «Зачем нам трихоплакс» Посмотреть видео |
 |
Алиса Левинсон «Репродуктивные технологии» Посмотреть видео |
 |
Михаил Карташов «Уроки Эболы» Посмотреть видео |
 |
Анна Новиковская»До начала зверей: история наших предков до того, как они стали млекопитающими» Посмотреть видео |
 |
Анастасия Проскурякова «Кито-парнокопытные в воде и на суше» Посмотреть видео |
 |
Екатерина Сколотнева «Грибы» Посмотреть видео |
 |
День Дарвина 2021 Посмотреть видео |
 |
Антон Цыбко «Дивный новый мир психоделической терапии» Посмотреть видео |
 |
Алексей Маслов «Летучие мыши и где они обитают в Сибири» Посмотреть видео |
 |
Маргарита Романенко «Вакцины от КОВИД19: перед выбором»» Посмотреть видео |
 |
Михаил Цыганов «Генетика метаморфоза» Посмотреть видео |
 |
Дмитрий Березняков «Лучшее, конечно, вперед: основы научного оптимизма» Посмотреть видео |
 |
Елена Землянская «Системный анализ экспрессии генов: что скрывается под верхушкой айсберга?» Посмотреть видео |
 |
Илья Акбердин «Эпидемиология COVID 19» Посмотреть видео |
 |
Alexander Suh (Uppsala University) «Evolution of repetitive elements on sex-specific and germline-restricted chromosomes of birds» Посмотреть видео |
 |
Владимир Филоненко «Будни и праздники переводчика» Посмотреть видео |
 |
Елена Шнайдер «Сокол балобан: популяционная биология и охрана» Посмотреть видео |
 |
Алла Красикова «Хромосомы типа ламповых щеток в свете геномики» Посмотреть видео |
 |
Иван Полторацкий «Эволюция русской поэзии» Посмотреть видео |
 |
Михаил Карташов Антон Николенко Нариман Баттулин Иван Полторацкий Дмитрий Березняков Ольга Ечевская Все нобелевские премии 2020 в одной лекции Посмотреть видео |
 |
Маргарита Романенко «Вакцина нашей надежды» Посмотреть видео |
 |
Сергей Мурсалимов «Пластиды: 2 миллиарда лет рабства» Посмотреть видео |
 |
Маргарита Романенко»Тот самый вирус: все что вы хотели знать о COVID19, но стеснялись спросить» Посмотреть видео |
 |
Ирина Захарова Гипоксия и плюрипотентность Посмотреть видео |
 |
Дмитрий Константинов «Как растения c бактериями азот усваивали» Посмотреть видео |
 |
Татьяна Бабочкина «Фетальный химеризм» Посмотреть видео |
 |
Ольга Ечевская. «Дистанционное образование: до, после и вместо карантина» Посмотреть видео |
 |
Алина Дресвянникова «Лист» |
 |
Любовь Малиновская «Кот» |
 |
Светлана Фёдорова «Танцуют все! Миграция клеток в организме» Посмотреть видео |
 |
С. Шарапов. Генетический контроль гликозилирования Посмотреть видео |
 |
Дмитрий Ланшаков «Голова профессора Доуэля» |
 |
Emma Teeling «The secrets of the bat genome» Посмотреть видео |
 |
Mike Bruford (Cardif University, UK) «Population genomics and conservation of genetic diversity: how wild species can inform livestock and back again» |
 |
Вадим Климонтов Эволюционная ловушка: сахарный диабет Посмотреть видео |
 |
День Дарвина 2020 |
 |
Алексей Кораблёв «Мышь» Посмотреть видео |
 |
Александр Пилипенко «Происхождение и эволюция человека: новое и хорошо забытое старое» Посмотреть видео |
 |
Н.Б.Рубцов «Геном человека в раннем развитии» Посмотреть видео |
 |
Елизавета Куликова «Шизофрения:вымысел и реальность» Посмотреть видео |
 |
Елена Воропаева «Потеря гетерозиготности и канцерогенез» Посмотреть видео |
 |
Евгений Березиков Червь Macrostomum lignano и секреты регенерации и долголетия Посмотреть видео |
 |
Марина Зыцарь «История болезни: Наследственная глухота» Посмотреть видео |
 |
Дмитрий Константинов «Фотосинтез у паразитических растений» Посмотреть видео |
 |
О.А.Донских (НГУ) «Поэзия у колыбели науки: избранные страницы интеллектуальной истории» Посмотреть видео |
 |
Никита Торгунаков «Мейотический драйв: преступление и наказание» Посмотреть видео |
 |
Все Нобелевские премии 2019 года в одной лекции Посмотреть видео |
 |
Юлия Позднякова «СМИ для учёных: кто, кого и как (цитирует)» Посмотреть видео |
 |
Степан Белякин Фантастический геномный импринтинг у насекомых Посмотреть видео |
 |
Антон Богомолов 256 оттенков серого. Распознавание образов. Посмотреть видео |
 |
Luis Garcia «Tricyclo-DNA for systemic splice switching approaches». Посмотреть видео |
 |
В.Г. Розин «Золотое сечение» Посмотреть видео |
 |
Е. Бражник, Н. Попова «Трансплантация костного мозга» Посмотреть видео |
 |
Денис Ларкин Сравнительная геномика того, чего никогда не было и что из этого получилось Посмотреть видео |
 |
Анна Кукекова «Геном лисицы» Посмотреть видео |
 |
Татьяна Шнайдер «Мини-органы» Посмотреть видео |
 |
Александр Пилипенко «Новости эволюции человечества». Посмотреть видео |
 |
Дмитрий Березняков «Теория революции» Посмотреть видео |
 |
Полина Белокопытова «Машинное обучение в биологии и медицине» Посмотреть видео |
 |
Елизавета Орлова «Эволюция иммунной системы» Посмотреть видео |
 |
Антона Цыбко «Нейротрофические факторы в терапии болезни Паркинсона» Посмотреть видео |
 |
Оксана Волкова «Кодирующие длинные некодирующие РНК» Посмотреть видео |
 |
Михаил Матыцин «Игры — происхождение и эволюция» Посмотреть видео |
 |
Вадим Нимаев «Акведуки организма: Прошлое и настоящее лимфатической системы» Посмотреть видео |
 |
Любовь Малиновская «История ДНК, которой НЕТ в соматических клетках» Посмотреть видео |
 |
Дмитрий Гражданкин «Филогенетические джунгли докембрия» Посмотреть видео |
 |
София Хантемирова «Пищевая зависимость» Загрузить презентацию (pdf) Посмотреть видео |
 |
Станислав Дрёмов «1000 лет одиночества: генетическая история нивхов» Загрузить презентацию (pdf) |
 |
День Дарвина 2019 Павел Сальников «Креатив на креативе: вопиющие примеры сомнительного дизайна организмов» Павел Никулин «Последний дарвинист» Премьера фильма «После прочтения жить» |
 |
Федор Колпаков «Регуляция транскрипции: эхо минувшей войны»Загрузить презентацию (pdf) Посмотреть видео |
 |
Prof. J. Wilson «Big and Clever: inbreeding, fertility and health» Посмотреть видео |
 |
Ксения Стрыгина «Эволюция генома растений» Загрузить презентацию (pdf) Посмотреть видео |
|
Татьяна Шнайдер «Мини-органы» Посмотреть видео |
|
Александр Пилипенко «Новости эволюции человечества». Посмотреть видео |
|
Дмитрий Березняков «Теория революции» Посмотреть видео |
|
Полина Белокопытова «Машинное обучение в биологии и медицине» Посмотреть видео |
|
Елизавета Орлова «Эволюция иммунной системы» Посмотреть видео |
|
Антона Цыбко «Нейротрофические факторы в терапии болезни Паркинсона» Посмотреть видео |
|
Оксана Волкова «Кодирующие длинные некодирующие РНК» Посмотреть видео |
|
Михаил Матыцин «Игры — происхождение и эволюция» Посмотреть видео |
|
Вадим Нимаев «Акведуки организма: Прошлое и настоящее лимфатической системы» Посмотреть видео |
|
Любовь Малиновская «История ДНК, которой НЕТ в соматических клетках» Посмотреть видео |
|
Дмитрий Гражданкин «Филогенетические джунгли докембрия» Посмотреть видео |
|
София Хантемирова «Пищевая зависимость» Загрузить презентацию (pdf) Посмотреть видео |
|
Станислав Дрёмов «1000 лет одиночества: генетическая история нивхов» Загрузить презентацию (pdf) |
|
День Дарвина 2019 Павел Сальников «Креатив на креативе: вопиющие примеры сомнительного дизайна организмов» Павел Никулин «Последний дарвинист» Премьера фильма «После прочтения жить» |
|
Федор Колпаков «Регуляция транскрипции: эхо минувшей войны»Загрузить презентацию (pdf) Посмотреть видео |
|
Prof. J. Wilson «Big and Clever: inbreeding, fertility and health» Посмотреть видео |
 |
Ксения Стрыгина «Эволюция генома растений» Загрузить презентацию (pdf) Посмотреть видео |
 |
Татьяна Шнайдер «Искусство искусственных органов» Посмотреть видео |
 |
Нариман Баттулин «Микроскопом по гвоздю! Негенетические функции генома» Посмотреть видео |
 |
Александр Пилипенко «Скифы и другие люди» Посмотреть видео |
 |
Вениамин Фишман «3D-мета-гено-пере-GWAS: метод Hi-C в биологии и медицинской генетике» Загрузить презентацию (pptx) Посмотреть видео |
 |
Т.Ф.Чалкова «Этика и практика научных журналов» Загрузить презентацию (pdf) |
 |
Елена Строкова «Сколиоз: история болезни» Загрузить презентацию (pdf) Посмотреть видео |
 |
Александр Шевченко «Дозовая компенсация» Загрузить презентацию (pdf) Посмотреть видео |
 |
Анна Дружкова «Очень древняя ДНК» Посмотреть видео |
 |
Игорь Косенко «Мир юрского периода» Загрузить презентацию (pdf) Посмотреть видео |
 |
Наталия Аульченко Мотивационные и рекомендательные письма Загрузить презентацию (pdf) |
 |
Ульяна Архипова «Лекарства: от идеи до аптеки» |
 |
Почти все Нобелевские премии 2018 года в одной лекции Посмотреть видео |
 |
Максим Королёв «Ревматоидный артрит» Посмотреть видео |
 |
Яков Цепилов «Генетика боли в спине» Посмотреть видео |
 |
Александр Кель «Сюрреализм генома» Загрузить презентацию (pdf) Посмотреть видео |
 |
Ирина Мухамедшина «Выражение эмоций у человека и животных» Загрузить презентацию (pdf) Посмотреть видео |
 |
Александр Бакланов «История болезни: ожирение» |
 |
Михаил Тюменцев «Нейродегенерация без границ: клубок протеинопатий» Загрузить презентацию (pdf) Посмотреть видео |
 |
С.Л.Николаев «География русских диалектов» Диалектологический атлас Посмотреть видео |
 |
Александр Пилипенко «Денисовцы, неандертальцы и другие люди» Загрузить презентацию (pdf) Посмотреть видео |
 |
Кирилл Устьянцев «Мобильные элементы и эволюция геномов» Загрузить презентацию (pdf) Посмотреть видео |
 |
Яна Сизенцова: Имплантация зубов: вчера, сегодня, завтра Загрузить презентацию (pdf) Посмотреть видео |
 |
Анастасия Юнусова «Методы изучения судьбы отдельных клеток» Посмотреть видео |
 |
Анна Смирнова «Фактор, индуцируемый гипоксией: враг или помощник» Загрузить презентацию (pdf) Посмотреть видео |
 |
Леонид Климов «История болезни: гипертония» Посмотреть видео |
 |
Мария Орлова. Дело «хозяйское» От чего зависит зараженность паразитами? Загрузить презентацию (pdf) Посмотреть видео |
 |
День Дарвина в ИЦИГ 2018 Stand-up Ларисы Мейстер: «Псы не то, чем кажутся» Поэма Павла Никулина «Идея» Клип Евгения Тийса «Дарвин рисует дождь» Балетный детектив «Дело о пляшущих вьюрках» Посмотреть видео |
 |
С. Герасимова. Зачем CRISPR/Cas каждому из нас Посмотреть видео |
 |
Анастасия Глаголева «Меланины — секретные пигменты растений» Загрузить презентацию (pdf) Посмотреть видео |
 |
Татьяна Фролова «Фуфломицин: инструкция по применению» Загрузить презентацию (pdf) Посмотреть видео |
 |
Михаил Чернов «Опухоли мозга: диагноз и лечение» Загрузить презентацию (pdf) Посмотреть видео |
 |
Хулио Фернандес «Омика раковых клеток» Посмотреть видео |
 |
Яков Цепилов «ПГАА: что делать дальше» Посмотреть видео |
 |
Алексей Катохин «Эволюция жизненных форм паразитов» Загрузить презентацию (pdf) Посмотреть видео |
 |
Татьяна Колесникова (ИМКБ СОРАН) «Эпигенетика и полифенизм насекомых» Загрузить презентацию (pdf) Посмотреть видео |
 |
Михаил Тюменцев «Митохондриальные болезни» Загрузить презентацию (pdf) Посмотреть видео |
 |
Галина Азаркина «Пауки-скакунчики: от морфологии до молекулярной генетики» Посмотреть видео |
 |
«Пять нобелевских премий 2017 года в одной лекции»» Посмотреть видео |
 |
Мария Побединцева «Мифы и факты об осетровых рыбах» Загрузить презентацию (pdf) Посмотреть видео |
 |
Алексей Мостович «Молекулярная электроника» Посмотреть видео |
 |
Владимир Трифонов «Полногеномные дупликации и эволюция позвоночных» Загрузить презентацию (pdf) Посмотреть видео |
 |
Дарья Новикова «Что делать со списком дифференциально экспрессируемых генов» Загрузить презентацию (pdf) Посмотреть видео |
 |
Беляевские чтения. P. Mormede, E.E. Terenina. Molecular genetics of the adrenocortical axis and breeding for robustness Загрузить презентацию (pdf) Посмотреть видео |
 |
Беляевские чтения. Д.М. Ларкин. Структура популяций и следы селекции в геномах пород КРС, разводимых в России Загрузить презентацию (pdf) Посмотреть видео |
 |
Беляевские чтения. S.J. O’Brien. The taming of the cat Загрузить презентацию (pdf) Посмотреть видео |
 |
Беляевские чтения. А.О. Рувинский. Гены и свет: много лет спустя Загрузить презентацию (pdf) Посмотреть видео |
 |
Беляевские чтения. А.О. Рувинский о Д.К.Беляевe Загрузить презентацию (pdf) |
 |
Беляевские чтения. H.V. Westerhoff. Principles of systems biology and Dmitri Belyaev’s co-selection of traits Загрузить презентацию (pdf) Посмотреть видео |
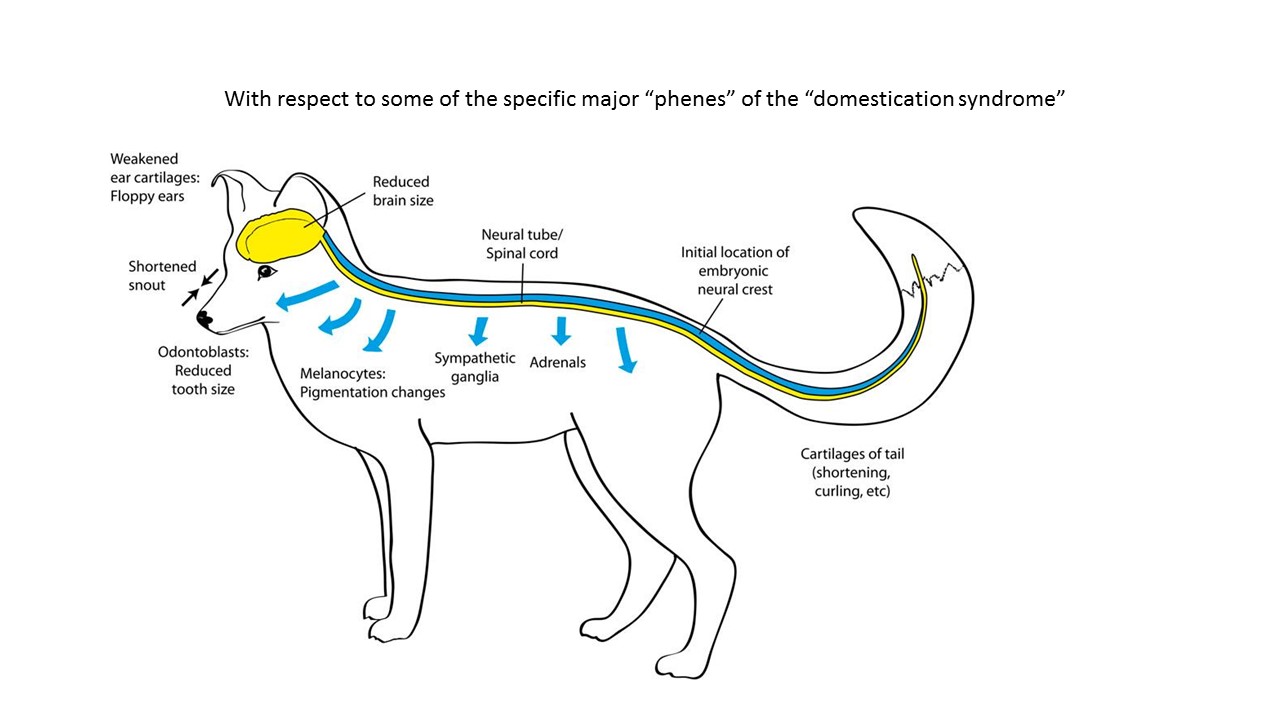 |
Беляевские чтения. A.S. Wilkins. Examining two hypotheses on the “domestication syndrome” Загрузить презентацию (pdf) Посмотреть видео |
 |
Беляевские чтения. И.А. Захаров-Гезехус, Ю.А. Столповский. Сохранение генофондов доместицированных видов животных Загрузить презентацию (pdf) Посмотреть видео |
 |
Беляевские чтения. В.С. Баранов. Эволюция предиктивной медицины Загрузить презентацию (pdf) Посмотреть видео |
 |
Андрей Кузьминов «Лаборатория как среда обитания» |
 |
Екатерина Трифонова «Аутизм: информация к размышлению» Загрузить презентацию (pdf) Посмотреть видео |
 |
Павел Никулин «Вирусы, которые мы приручили» |
 |
Станислав Дрёмов «Выжившие: генетическая история эскимосов» Загрузить презентацию (pdf) |
 |
Ольга Голубкова «Нежить» и «неведомая сила» Посмотреть видео |
 |
Содбо Шарапов «Неуловимый эпистаз» Загрузить презентацию (pdf) |
 |
Евгений Симонов «Позвоночные юга Сибири» |
 |
Наталья Торгашева «Порочный круг болезни Альцгеймера» Загрузить презентацию (pdf) Посмотреть видео |
 |
Софья Герасимова «Страсти по ГМО» Загрузить презентацию (pdf) Посмотреть видео |
 |
Алексей Маслов «Пещерная жизнь» Загрузить презентацию (pdf) Посмотреть видео |
 |
Александр Кононов «Чужие: генетика инвазивных популяций» Загрузить презентацию (pdf) Посмотреть видео |
 |
Пять Нобелевских премий 2016 года в одной лекции: сборная публичная лекция Алексея Миронова (ИФП) — премия по физике, Евгения Мостовича (НГУ) — премия по химии, Константина Орищенко (ИЦИГ) — премия по физиологии и медицине, Ольги Ечевской (НГУ) — премия по экономике, Ивана Полторацкого (НГУ) — премия по литературе Посмотреть видео |
 |
Александра Клименко «Бактерии и фаги: сводки с фронта и тыла» Загрузить презентацию (pdf) Посмотреть видео |
 |
Проф. А.Д.Долгов «От большого взрыва до…» Посмотреть видео |
 |
Тропа предков Посмотреть фоторепортаж |
 |
Michel Georges «The impact of genomics in animal breeding» Посмотреть видео |
 |
Мария Юдина «Паразиты паразитов» Загрузить презентацию (pdf) |
 |
Илья Захаров «Генетика когнитивных способностей» Загрузить презентацию (pdf) Посмотреть видео |
 |
Андрей Табарев (ИАЭТ СО РАН) «Неолитическая революция и неолитизация» Загрузить презентацию (pdf) Посмотреть видео |
 |
Алиса Серяпина «Эвтаназия лабораторных животных: как и зачем»» Посмотреть видео |
 |
Ольга Ечевская «Экономика и социология счастья» Посмотреть видео |
 |
Андрей Каява «Темная материя протеома: пространственная структура неглобулярных белков». Загрузить презентацию (pdf) Посмотреть видео |
 |
Татьяна Шнайдер «Уходя, гасите ген: методы управления экспрессией» Загрузить презентацию (pdf) Посмотреть видео |
 |
Проф. Д.В.Гражданкин (ИНГГ) «Экосистемы Эдиакария» Посмотреть видео |
 |
Надежда Белоногова «Реквием по уровню значимости» Загрузить презентацию (pdf) |
 |
Александр Пилипенко «Гены забытых предков» Загрузить презентацию (pdf) Посмотреть видео |
 |
Мария Львова «Гематофаги: вампиры внутри нас» |
 |
Проф. А.Д. Долгов (ИЯФ) «Гравитационные волны» Загрузить презентацию (pdf) Посмотреть видео |
 |
Андрей Кечин «Туберкулёз: один раз — и навсегда» Посмотреть видео |
 |
Дина Малькеева «Вначале было ядро: происхождение эукариот» Посмотреть видео |
 |
ДЕНЬ ДАРВИНА 1. Лекция И.Полторацкого (НГУ) «Литературный стиль Дарвина» 2. Оратория П.Никулина «Дарвин-супергерой» 3. Премьера фильма «Дарвин вернулся» |
 |
Антон Нижников (кафедра генетики и биотехнологии СПбГУ) «Амилоиды и прионы: от патогенеза к функции» Загрузить презентацию (pdf) |
 |
Анастасия Юнусова «Терапия стволовыми клетками: где мы сейчас?» |
 |
Мария Пахарукова «Паразит — хозяин: стратегия выживания» |
 |
Алла Брянская «Мир микробов: новое или неизвестное? |
 |
Проф. М.С. Гельфанд «Регуляция транскрипции у бактерий: эволюция и ко-эволюция»» |
 |
Михаил Цыганов «Естественная история малярии» |
 |
Ильяс Джетыбаев «Определение пола у насекомых: всё не так просто как вы думали» |
 |
Dr. Ivo Grosse (Martin-Luther University, Germany) «Phylotranscriptomic hourglasses of plant and animal embryogenesis» |
 |
Светлана Федорова «И все-таки они вертятся! Жгутики и реснички – биологические моторы». |
 |
Алексей Дорошков «Эволюция регенерации». |
 |
Вениамин Фишман «Генетика старения». |
 |
Денис Ларкин «Стая птичьих геномов» |
 |
Юрий Илинский «Простейшие: всё сложно» |
 |
Анна Гриневич (Ин-т филологии СОРАН) «Эволюция фольклорных сюжетов» |
 |
Елена Кизилова «Откуда руки растут: морфогенез конечностей» |
 |
С.Л.Николаев (Ин-т славяноведения РАН) «Родство языков» |
 |
Екатерина Новосёлова «Светлые и темные полосы морфогенеза» |
 |
Prof. Michel Georges (University of Liege, Belgium) «Unexpectedly high mutation rate at early cleavage cell divisions» |
 |
Дина Малькеева «Белки теплового шока: структура и функции» |
 |
Александр Пилипенко «Не ждали нас, а мы пришли: молекулярная история человечества». |
 |
Ольга Сайк «Генные сети: что в них можно поймать?» |
 |
Виктория Миронова «Почему выживают нокауты? История одного растения» |
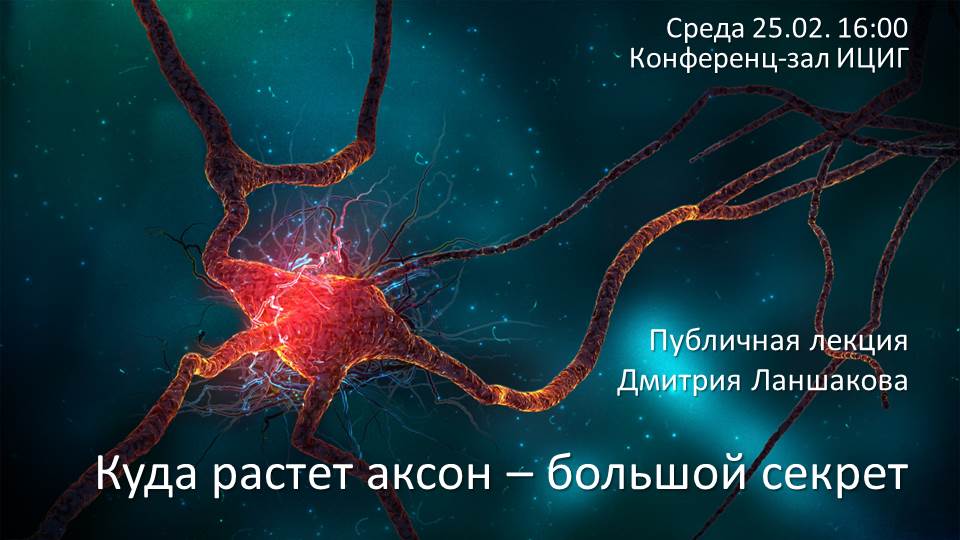 |
Дмитрий Ланшаков «Куда растет аксон — большой секрет» |
 |
Михаил Помазной «Паразит — хозяин: битва на молекулярном уровне» |
 |
. День Дарвина 2015 Посмотреть видео |
 |
. День Дарвина 2015 Посмотреть видео: 00-35 мин |
 |
. День Дарвина 2015 Посмотреть видео: 35-39 мин |
 |
. День Дарвина 2015 Посмотреть видео: 39-59 мин |
 |
Надежда Белоногова «Количественная генетика» |
 |
В.М. Ефимов «Зачем биологу биостатистика» |
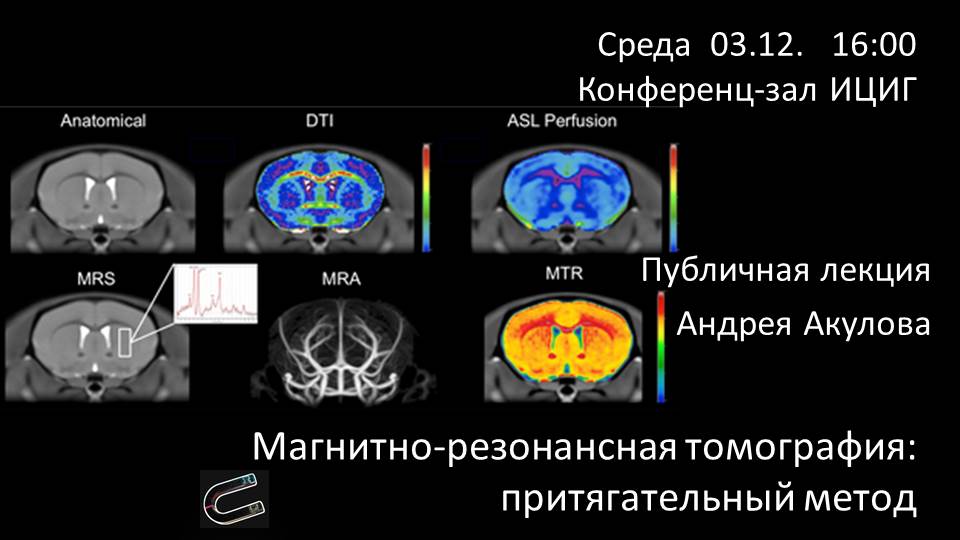 |
Андрей Акулов «Магнитно-резонансная томография: притягательный метод» |
 |
П.Д.Лисачев «Поиски карты: Нобелевская премия по физиологии и медицине 2014» |
 |
Светлана Фоминых «Наука и искусство публичных выступлений» |
 |
Дина Логинова «Мейоз амфигаплоидов: курс на выживание» |
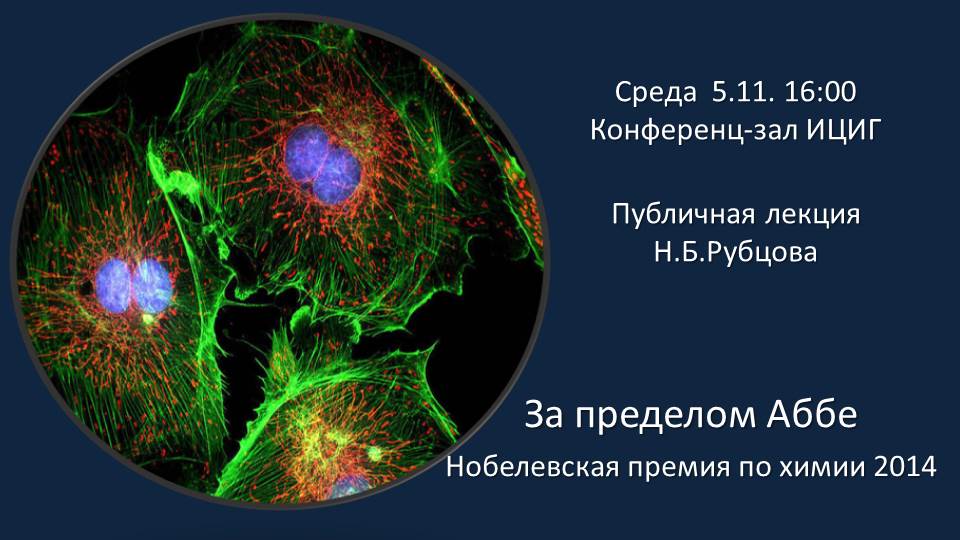 |
Н.Б. Рубцов «За пределом Аббе: Нобелевская премия по химии 2014″» |
 |
Елизавета Куликова «Депрессия: состояние души или нарушение мозга?» |
 |
Н.В. Рубцова «Аспирантура в ИЦиГ: кем быть и что делать?» |
 |
Дарья Андреюшкова «Геномы птиц» |
 |
Проф. Е.И. Рогаев «Молекулярные основы болезни Альцгеймера» |
 |
Артем Лисачев «Взлет и падение Y хромосомы» |
 |
Dr. Christian Gieger (Helmholtz Center, Munich) «Genetic and metabolic atlas helps to fight disease»
Загрузить презентацию (pdf)
Посмотреть видео |
 |
Нариман Баттулин «Ctrl+C + Ctrl+V или все, что вы хотели знать о клонировании» |
 |
Татьяна Колесникова «Эпигенетика. Новая парадигма?» |
 |
Анна Дружкова «Геномика псов(ых)» |
 |
Вениамин Фишман «Животный магнетизм» |
 |
Юрий Аульченко «Блеск и нищета предиктивной медицины или история болезни, предсказанной заранее» |
 |
Александр Пилипенко «Происхождение и эволюция человека: ещё год спустя» |
 |
Д.А. Жуков (ИФ им. И.П.Павлова) «Активная стратегия поведения как фактор риска депрессии» |
 |
Маргарита Тарасова «Вакцины в меняющемся мире» |
 |
Елена Хлесткина «Геном пшеницы: конструктор в руках исследователей» |
 |
Константин Орищенко «Геномная инженерия: возможности и перспективы» |
 |
День Дарвина 2014: П. Бородин «На поколение дальше от обезьяны» |
 |
День Дарвина 2014: П. Никулин «Поэтический Дарвин» |
 |
День Дарвина 2014: Студенты 4 курса НГУ |
 |
Федор Кондрашов «Патогенные мутации в свете эволюции» |
 |
Академик В.К. Шумный «Этюды по истории генетики» |
 |
Павел Бородин «Происхождение видов полтора века спустя» |
 |
Олеся Шоева «Пигменты растений» |
 |
Александр Смирнов «Происхождение репликации» |
 |
Анатолий Федотов (ИВТ СОРАН) «Модели будущей погоды» |
 |
Илья Любечанский (ИСИЭЖ СО РАН) «Глобальное изменение климата: кто виноват?» |
 |
Павел Никулин «Вироиды и пандоравирусы: парадокс сложности» |
 |
Сергей Седых (ИХБФМ) «Клеточная почта: везикулярный трафик |
 |
Павел Кроковный (ИЯФ) «Бозон Хиггса» |
 |
Артем Лисачев «Происхождение однополых видов путем гибридизации, или сохранение клональных форм в борьбе за жизнь» |
 |
Мария Химена Фернандес Гомес «Эволюция под землей: история туко-туко» |
 |
Дмитрий Ланшаков «Дежавю и другие манипуляции с мышиным сознанием» |
 |
Антон Струнов «Wolbachia – внутриклеточный иммигрант» |
 |
Christian Gieger and Janina Ried «Refined analysis strategies for genome-wide association analysis to understand diseases in humans» |
 |
Вячеслав Власов «Рак и лекарства: есть ли свет в конце туннеля?» |
 |
Елена Кизилова «Реальность химеры» |
 |
Сергей Коваленко (ИМББ СО РАМН) «Рак — болезнь генома. Возможны ли лекарства?» |
 |
Маргарита Тарасова (НГУ) «Вирусы против рака: проблемы и решения» |
 |
Александр Пилипенко «Происхождение и эволюция человека: полтора года спустя» |
 |
Софья Пантелеева «Эволюция поведенческих стереотипов» |
 |
Александр Савостьянов «Как наш мозг понимает то, что видит глаз?»Загрузить презентацию (pdf) Посмотреть видео |
 |
Сергей Шеховцов «Молекулярная филогения Metazoa» |
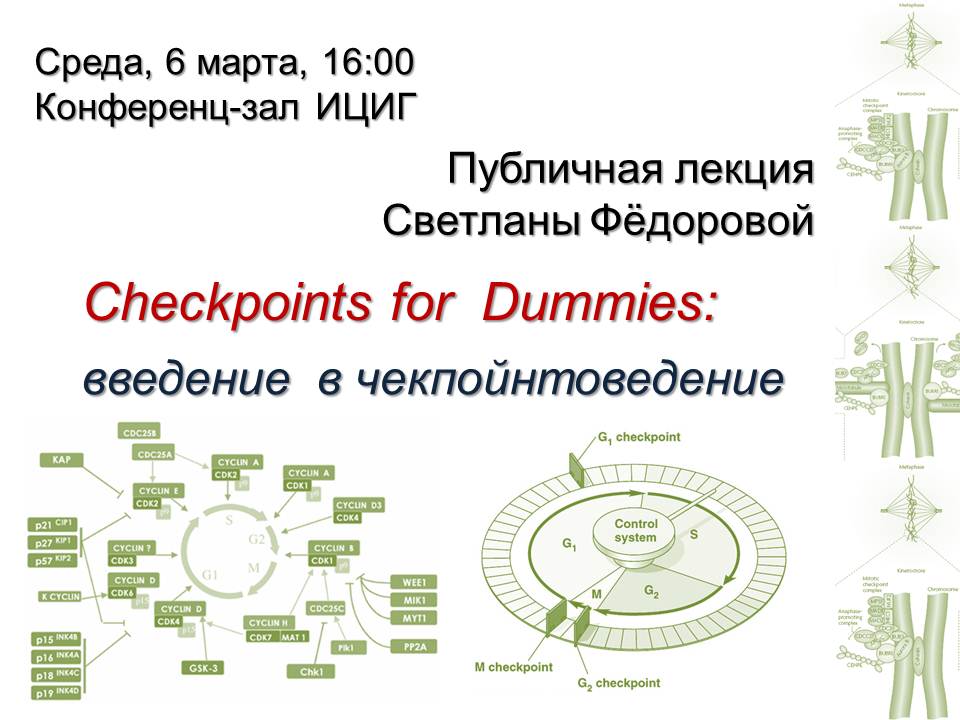 |
Светлана Федорова “Checkpoints for Dummies:введение в чекпойнтоведение”
Загрузить презентацию (pdf) |
 |
Геннадий Васильев “Как собрать ген из букв: несколько практических советов для начинающих” |
 |
Александр Иляскин «Плазматическая мембрана: государственная граница клетки» |
 |
Дмитрий Щербаков (Лимнологический институт) «Эволюция фауны Байкала» |
 |
Eulogy to Charles Darwin on the occasion of his 204 birthday written by Pavel Nikulin, BSc, and delivered at the Darwin’s Day celebration at the Institute of Cytology and Genetics |
 |
Дарвин в альтернативной реальности (пьеса в 8 действиях) |
 |
Татьяна Шнайдер «Многодетищный отец» |
 |
Евгений Тийс «Как найти статьи о том, как найти статьи?» |
 |
Ксения Сорокина «Про зеленую химию» |
 |
Дмитрий Гражданкин (Институт нефтегазовой геологии и геофизики СОРАН) «Запретные плоды Эдиакарского сада» |
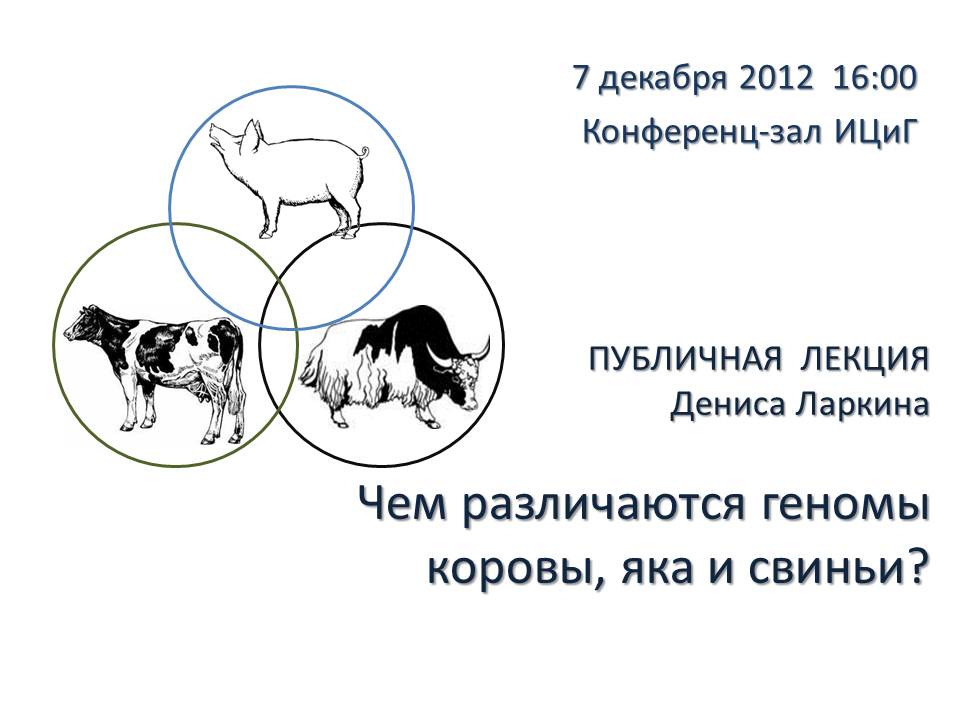 |
Денис Ларкин «Чем различаются геномы коровы, яка и свиньи?» |
 |
Надежда Баттулина «Взгляд в глубину: эволюция глаза» |
| Ирина Сормачева «Горизонтальный перенос у эукариот: we are not only what we eat …» | |
 |
Проф. В.Н. Анисимов «Успехи геронтологии: есть ли у нас таблетка от старости?» |
 |
Иван Бурков «РНК: Транскрипция — это только начало» |
 |
Нариман Баттулин «Геномный импринтинг» |
 |
Дмитрий Афонников «Эволюционная биоинформатика и реконструкция филогении» |
 |
Проф. Ю.Ф.Богданов «Особенности организации мейотической хромосомы» |
 |
John Parker «A long way to The Origin» |
 |
Юлия Минина «Теломеры: The last but not the least» |
 |
Проф. В.И. Тельнов (ИЯФ) «Темные материя и энергия во вселенной» |
 |
Елена Хлесткина «Легко ли быть аллополиплоидом» |
 |
Дарья Базовкина «Молекулярная генетика поведения грызунов» |
 |
Алексей Мензоров «Эмбриональные стволовые клетки, которых не существует» |
 |
Виктория Миронова «Основы теории морфогенеза» |
 |
Как делать доклады в PowerPoint |
 |
День Дарвина-2012. Артем Лисачев «Дарвин: перезагрузка»
Загрузить презентацию Премьерный показ фильма «Чарльз»
|
 |
Елена Кизилова «Как стать многоклеточным? Индивидуальный выбор млекопитающих» |
 |
Алла Брянская «Жизнь без ядра» |
 |
Анна Торгашева «Мейоз»
Загрузить презентацию (pdf) |
 |
Юрий Орлов «Геном человека» |
 |
Юрий Илинский «Симбиоз и симбиогенез» |
 |
Владимир Бабенко «Сплайсинг» |
 |
Сергей Лашин «Биологическая эволюция — вчера, сегодня, никогда?» |
 |
Александр Пилипенко «Происхождение и эволюция человека» |
 |
Илья А. Захаров-Гезехус «Красота генетики»
«Потомки Чингизхана» Загрузить презентацию 2 (pdf) |
 |
Елена Дементьева «Дозовая компенсация» |
| Нариман Баттулин «Генетический контроль индивидуального развития» | |
 |
Как делать доклады в PowerPoint |
 |
Наталья Губанова «Клеточная смерть в подробностях» |
| Ильяс Джетыбаев «Эволюция кариотипов» | |
 |
Елена Хлесткина «Молекулярные методы анализа» |
 |
Иван Бурков «Трансгенез: вчера сегодня, завтра» |
 |
Юрий Аульченко «Статистическая генетика сложных признаков человека» |
 |
Ксения Головнина «Повторяющиеся элементы» |